
![]()
![]()
This package is a toolkit for working with Biological Observation Matrix (BIOM) files. Features include reading/writing all ‘BIOM’ formats, rarefaction, alpha diversity, beta diversity (including ‘UniFrac’), summarizing counts by taxonomic level, subsetting, visualizations, and statistical analysis. All CPU intensive operations are written in C.
Reference material is available online at https://cmmr.github.io/rbiom/index.html
Source code can be found at https://github.com/cmmr/rbiom
The latest stable version can be installed from CRAN.
install.packages("pak")
pak::pak("rbiom")The development version is available on GitHub.
pak::pak("cmmr/rbiom")library(rbiom)
infile <- system.file(package = "rbiom", "extdata", "hmp50.bz2")
biom <- rarefy(infile)bdiv_ord_plot(biom, stat.by = "Body Site", facet.by = "Sex")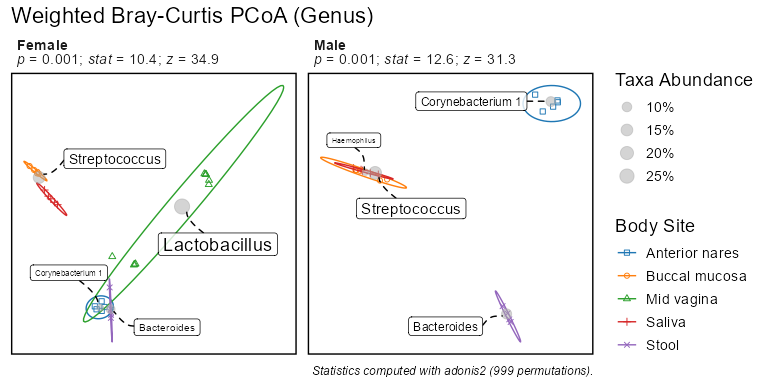
adiv_boxplot(biom, x = "Sex", adiv = c("otu", "shan"), stat.by = "Body Site")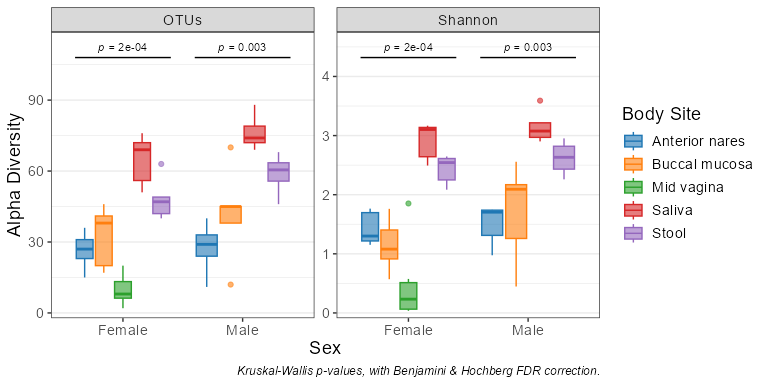
subset(biom, `Body Site` == 'Buccal mucosa') %>%
taxa_corrplot("Age", taxa = 2, layers = 'ptc', fit = 'lm', test = 'emtrends')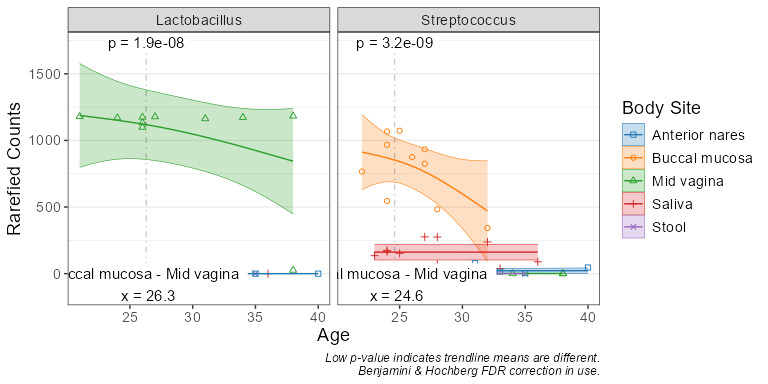
taxa_heatmap(biom, taxa = 10, tracks = c("body", "age"))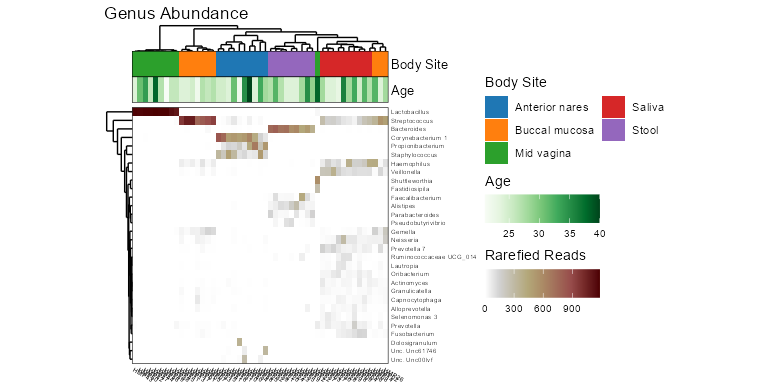
taxa_stacked(biom, rank = "Phylum")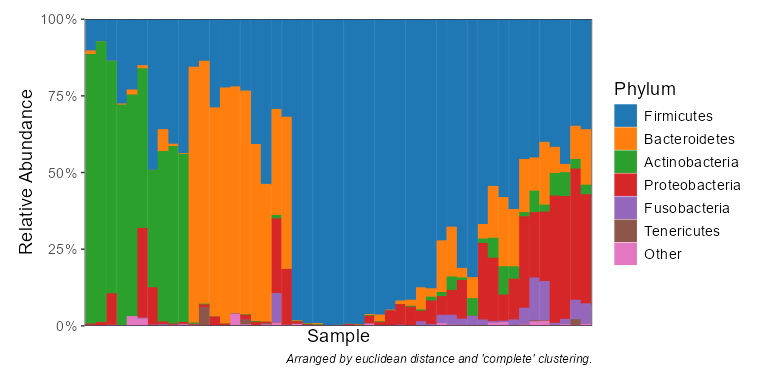
taxa_table(biom, 'Phylum')
#> # A tibble: 294 × 8
#> .rank .sample .taxa .abundance Age BMI `Body Site` Sex
#> <fct> <chr> <fct> <dbl> <dbl> <dbl> <fct> <fct>
#> 1 Phylum HMP01 Firmicutes 856 22 20 Buccal mucosa Female
#> 2 Phylum HMP01 Bacteroidetes 199 22 20 Buccal mucosa Female
#> 3 Phylum HMP01 Actinobacteria 16 22 20 Buccal mucosa Female
#> 4 Phylum HMP01 Proteobacteria 72 22 20 Buccal mucosa Female
#> 5 Phylum HMP01 Fusobacteria 32 22 20 Buccal mucosa Female
#> 6 Phylum HMP01 Tenericutes 0 22 20 Buccal mucosa Female
#> 7 Phylum HMP02 Firmicutes 803 24 23 Buccal mucosa Male
#> 8 Phylum HMP02 Bacteroidetes 192 24 23 Buccal mucosa Male
#> 9 Phylum HMP02 Actinobacteria 52 24 23 Buccal mucosa Male
#> 10 Phylum HMP02 Proteobacteria 96 24 23 Buccal mucosa Male
#> # ℹ 284 more rows